A DeepMind já conquistou uma série de vitórias, apresentando IAs que aprenderam a jogar uma variedade de jogos complexos com habilidades sobre-humanas, de Go e StarCraft a todo o catálogo da Atari. Mas Demis Hassabis, a figura pública e cofundador da DeepMind, sempre enfatizou que esses sucessos foram apenas trampolins para um objetivo maior: IA que realmente nos ajuda a entender o mundo.
Na última semana de novembro, a DeepMind e os organizadores da tradicional competição conhecida como Avaliação Crítica da Predição da Estrutura de Proteínas (em inglês, CASP) anunciaram uma IA que deve ter o enorme impacto que Hassabis tem procurado. A última versão do AlphaFold da DeepMind, o sistema de deep learning capaz de prever com precisão a estrutura das proteínas na largura de um átomo, resolveu um dos grandes desafios da biologia. “É o primeiro uso de IA para resolver um problema sério”, diz John Moult, da Universidade de Maryland, que lidera a equipe que administra o CASP.
Uma proteína é composta por um conjunto de aminoácidos que se dobra em muitas voltas e emaranhados complexos. Essa estrutura determina o que ela faz. E descobrir o que as proteínas fazem é a chave para compreender os mecanismos básicos da vida, quando funciona e quando não. Os esforços para desenvolver vacinas para a Covid-19 têm se concentrado na proteína spike do vírus, por exemplo. A maneira como o coronavírus se liga às células humanas depende da forma dessa proteína e daquelas do lado de fora dessas células. A spike é apenas uma proteína entre bilhões de todos os seres vivos; existem dezenas de milhares de tipos diferentes de proteínas apenas no corpo humano.
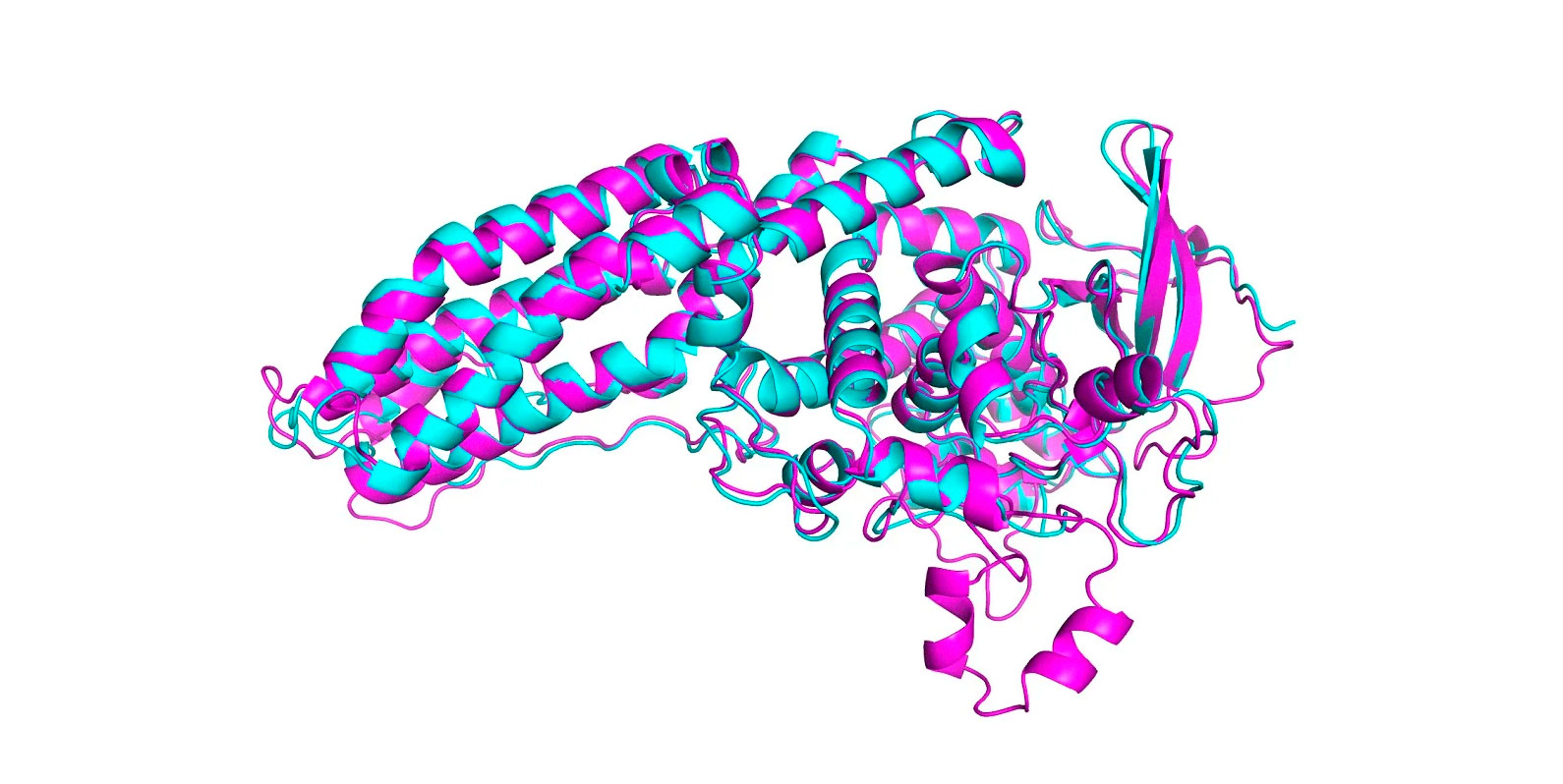
No CASP deste ano, AlphaFold previu a estrutura de dezenas de proteínas com uma margem de erro de apenas 1,6 angstroms, ou seja, 0,16 nanômetros, ou do tamanho de um átomo. Isso supera de longe todos os outros métodos computacionais e pela primeira vez corresponde à precisão das técnicas usadas no laboratório, como microscopia crio-eletrônica, ressonância magnética nuclear e cristalografia de raios-x. Essas técnicas são caras e lentas: podem levar centenas de milhares de dólares e anos de tentativa e erro para cada proteína. O AlphaFold pode encontrar a forma de uma proteína em poucos dias.
A descoberta pode ajudar os pesquisadores a desenvolver novos medicamentos e a compreender as doenças. No longo prazo, prever a estrutura da proteína também ajudará a projetar proteínas sintéticas, como enzimas que digerem resíduos ou produzem biocombustíveis. Os pesquisadores também estão explorando maneiras de criar proteínas sintéticas que aumentarão a produtividade das safras e tornarão os cultivos mais nutritivos.
“É um avanço muito substancial”, diz Mohammed AlQuraishi, biólogo de sistemas da Universidade de Columbia que desenvolveu seu próprio software para prever a estrutura de proteínas. “É algo que eu simplesmente não esperava que acontecesse tão rapidamente. É chocante, de certa forma”.
“Tem uma grande importância”, diz David Baker, chefe do Institute for Protein Design da Universidade de Washington, nos Estados Unidos, e líder da equipe por trás da Rosetta, um grupo de ferramentas de análise de proteínas. “É uma conquista incrível, como o que eles fizeram com o jogo Go”.
Números astronômicos
Identificar a estrutura de uma proteína é muito difícil. Para a maioria delas, os pesquisadores têm a sequência de aminoácidos da cadeia, mas não a forma contorcida em que se dobram. E normalmente há um número astronômico de formas possíveis para cada sequência. Os pesquisadores vêm lutando com o problema pelo menos desde os anos 1970, quando Christian Anfinsen ganhou o prêmio Nobel por mostrar que as sequências determinavam a estrutura.
O lançamento do CASP em 1994 deu um impulso ao campo. A cada dois anos, os organizadores lançam cerca de 100 sequências de aminoácidos para proteínas cujas formas foram identificadas em laboratório, mas ainda não publicadas. Dezenas de equipes de todo o mundo competem para encontrar a maneira correta de dobrá-las usando um software. Muitas das ferramentas desenvolvidas para CASP já são utilizadas por pesquisadores médicos. Mas o progresso ainda era lento, com duas décadas de avanços incrementais que falharam em produzir um atalho para o minucioso trabalho de laboratório.
O CASP teve o impacto que procurava quando a DeepMind entrou na competição em 2018 com sua primeira versão do AlphaFold. Ainda não tinha a precisão de um laboratório, mas deixou outras técnicas computacionais comendo poeira. Os pesquisadores tomaram nota: logo muitos estavam adaptando seus próprios sistemas para funcionar mais como o AlphaFold.
Este ano, mais da metade das inscrições usa alguma forma de deep learning, diz Moult. Como resultado, a precisão geral foi maior. O novo sistema de Baker, chamado trRosetta, usa algumas das ideias da DeepMind de 2018. Mas ainda veio um “segundo lugar muito distante do primeiro”, diz ele.
No CASP, os resultados são pontuados usando o que é conhecido como teste de distância global (em inglês, Global Distance Test ou GDT), que mede em uma escala de 0 a 100 quão próxima uma estrutura prevista está da forma real de uma proteína identificada em experimentos de laboratório. A versão mais recente do AlphaFold pontuou bem para todas as proteínas no desafio. Mas obteve uma pontuação de GDT acima de 90 para cerca de dois terços deles. Seu GDT para as proteínas mais difícies foi de 25 pontos mais altos do que a segunda melhor equipe, diz John Jumper, que lidera a equipe AlphaFold na DeepMind. Em 2018, a liderança girava em torno de seis pontos.
Uma pontuação acima de 90 significa que quaisquer diferenças entre a estrutura prevista e a real podem ser devidas a erros experimentais no laboratório, e não a uma falha no software. Isso também pode significar que a estrutura prevista é uma configuração alternativa válida para aquela identificada em laboratório, dentro da faixa de variação natural.
De acordo com Jumper, havia quatro proteínas na competição que os juízes independentes não tinham terminado de trabalhar no laboratório e as previsões de AlphaFold apontaram para as estruturas corretas.
AlQuraishi achou que os pesquisadores levariam 10 anos para passar dos resultados de 2018 da AlphaFold até os deste ano. Isso está perto do limite físico de quanta precisão você pode obter, diz ele. “Essas estruturas são fundamentalmente flexíveis. Não faz sentido falar sobre soluções muito abaixo disso”.
Peças de quebra-cabeças
AlphaFold se baseia no trabalho de centenas de pesquisadores em todo o mundo. DeepMind também contou com uma ampla gama de conhecimentos, reunindo uma equipe de biólogos, físicos e cientistas da computação. Detalhes de como funciona foram divulgados na última semana de novembro na conferência CASP e em um artigo revisado por pares em uma edição especial da revista Proteins que será lançada no próximo ano. Mas sabemos que ele usa uma forma de rede de atenção, uma técnica de deep learning que permite que uma IA seja treinada ao focar em partes de um problema maior. Jumper compara a abordagem a um quebra-cabeça: as peças são primeiro colocadas juntas separadamente antes de juntá-las em um todo.
A DeepMind treinou o AlphaFold em cerca de 170.000 proteínas retiradas do banco de dados de proteínas, um repositório público de sequências e estruturas. Ele comparou várias sequências no banco de dados e procurou pares de aminoácidos que muitas vezes acabam juntos em estruturas enoveladas. Em seguida, usa esses dados para adivinhar a distância entre pares de aminoácidos em estruturas que ainda não são conhecidas. Também é capaz de avaliar a precisão dessas suposições. O treinamento levou “algumas semanas”, usando uma capacidade de computação equivalente a entre 100 e 200 GPUs.
Dame Janet Thornton, do European Bioinformatics Institute em Cambridge, Reino Unido, trabalha com proteínas há 50 anos. “Esse problema existe há mais ou menos esse mesmo período”, disse ela em uma coletiva de imprensa em novembro. “Eu estava começando a achar que isso não seria resolvido durante a minha vida”.
Muitos medicamentos são projetados simulando sua estrutura molecular 3D e procurando maneiras de encaixar essas moléculas nas proteínas alvo. Claro, isso só pode ser feito se a estrutura dessas proteínas for conhecida. Este é o caso de apenas um quarto das cerca de 20.000 proteínas humanas, diz Thornton. Isso deixa 15.000 alvos de drogas inexplorados. “AlphaFold abrirá espaço para uma nova área de pesquisa”.
A DeepMind diz que planeja estudar leishmaniose, doença do sono e malária, todas doenças tropicais causadas por parasitas, porque estão ligadas a muitas estruturas de proteínas desconhecidas.
Uma desvantagem do AlphaFold é que ele é lento em comparação com as técnicas rivais. O sistema de AlQuraishi, que usa um algoritmo chamado rede geométrica recorrente (RGN), pode encontrar estruturas de proteínas um milhão de vezes mais rápido – encontrando resultados em segundos ao invés de dias. Suas previsões são menos precisas, mas para alguns aplicativos a velocidade é mais importante, diz ele.
Os pesquisadores agora estão esperando para descobrir exatamente como o AlphaFold funciona. “Assim que eles descreverem ao mundo como o fazem, mil outras formas de uso surgirão”, diz Baker. “As pessoas usarão para todos os tipos de coisas diferentes, coisas que não podemos imaginar agora”.
Mesmo um resultado menos preciso teria sido uma boa notícia para as pessoas que trabalham com enzimas ou bactérias, diz AlQuraishi: “Mas temos algo ainda melhor, com relevância imediata para aplicações farmacêuticas”.

